Il est essentiel de choisir une étiquette fabriquée à partir de matériaux de qualité pharmaceutique afin de garantir la sécurité des produits pharmaceutiques. Chez UPM Raflatac, nous garantissons la sécurité de nos étiquettes en utilisant des matériaux rigoureusement prétestés, conformes aux exigences réglementaires applicables pour l'usage auquel ils sont destinés et offrant un approvisionnement fiable sur le long terme.
Étiquettes pharmaceutiques et réglementation en vigueur
Étiquettes pharmaceutiques développées, formulées et fabriquées conformément à la réglementation en vigueur
Protéger les personnes
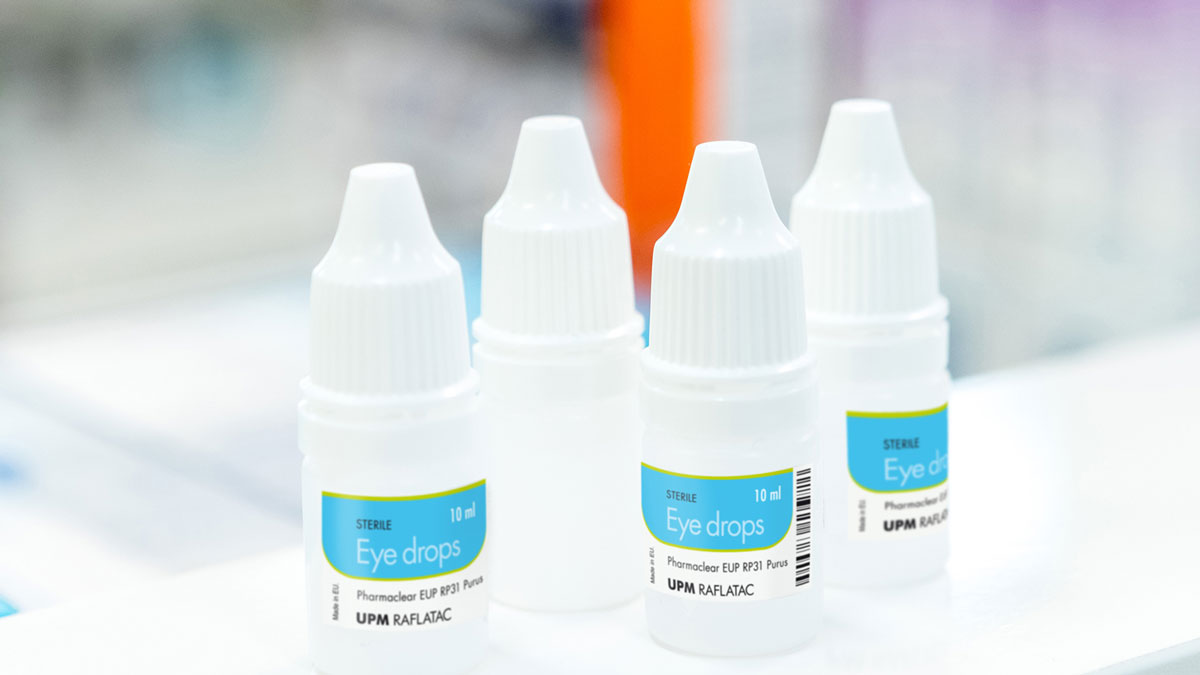
Étiquettes pharmaceutiques à faible migration
Les étiquettes jouent un rôle dans la protection de la sécurité en véhiculant des informations médicales cruciales. Elles peuvent également être source de préoccupation lorsque des produits pharmaceutiques nécessitent des matériaux à faible migration. Les médicaments liquides dans des emballages en plastique, tels que les gouttes oculaires, les sprays pour applications nasales ou les produits injectables, comptent parmi les produits pharmaceutiques qui peuvent nécessiter des étiquettes à faible lixiviabilité. Les matériaux pharmaceutiques que nous avons sélectionnés et notre technologie d'adhésif avancée ont été développés conformément aux directives du secteur, par exemple :
- La directive /CVMP/205/04 de l'EMA (Agence européenne des médicaments) sur les matériaux d'emballage primaire en plastique (2005)
- La directive de l'Agence américaine des produits alimentaires et médicamenteux (FDA, Food and Drug Administration) sur les systèmes de fermeture des contenants pour l'emballage des médicaments et des produits biologiques destinés à l'homme (1999)
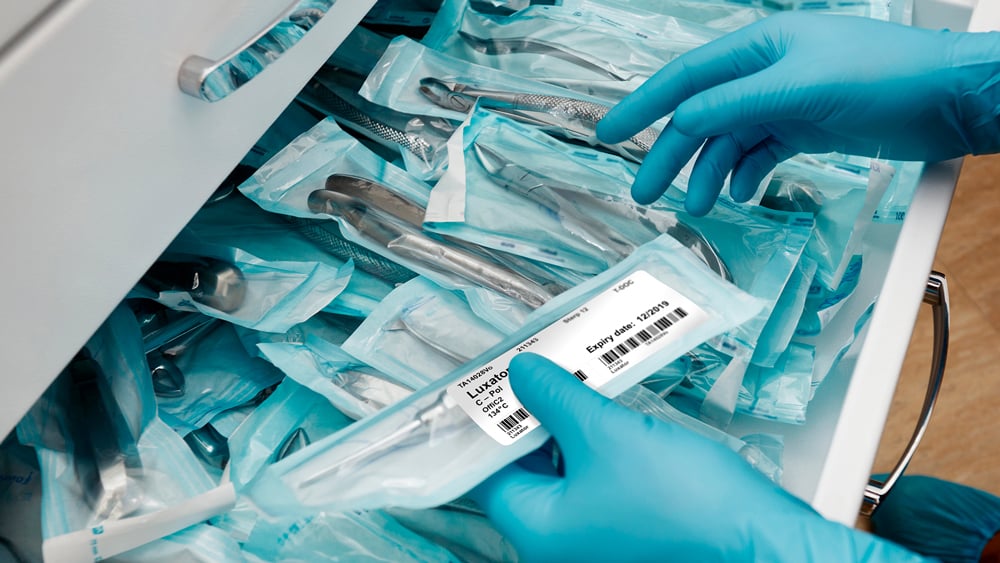
Réglementations relatives aux dispositifs médicaux
Les directives relatives aux dispositifs médicaux exigent que les systèmes d'impression et d'étiquetage soient compatibles avec les matériaux, les dispositifs et le traitement du système de protection stérile. Elles exigent également que les étiquettes restent intactes et lisibles jusqu'au point d'utilisation. L'étiquetage des dispositifs médicaux aide les patients ou les utilisateurs finaux à comprendre le dispositif, son fonctionnement, son nettoyage et son entretien. Les réglementations relatives aux dispositifs médicaux incluent :
- La nouvelle réglementation mondiale relative aux dispositifs médicaux, qui comprend l'adoption d'un système de sérialisation et d'identification unique des dispositifs médicaux (UDI) visant à marquer et identifier ces dispositifs au sein de la chaîne d'approvisionnement des produits de santé. L'une des dernières mises à jour réglementaires de l'UDI est la nouvelle réglementation européenne relative aux dispositifs médicaux [MDR], avec une date limite de mise en œuvre en mai 2020. Ce système est semblable à celui adopté par la FDA en 2013.
- Les réglementations sur les diagnostics in vitro (IVDR), qui constituent la base réglementaire pour la commercialisation, la mise à disposition et la mise en service de dispositifs médicaux de diagnostic in vitro sur le marché européen. La date limite de mise en œuvre des réglementations sur les diagnostics in vitro (IVDR) dans l'UE est fixée à mai 2022.
Directive relative aux médicaments falsifiés (2011/62/UE)
La Directive relative aux médicaments falsifiés (2011/62/UE) est applicable aux médicaments délivrés sur ordonnance et aux médicaments en vente libre, mais présentant un risque élevé. Conformément à la norme ISO 21976, « Emballage — Témoins d'effraction pour emballages de médicaments », ces produits possèdent des propriétés d'inviolabilité qui favorisent l'harmonisation et la mise en œuvre de caractéristiques anti-fraude pour les emballages de produits médicaux dans le monde entier.
Réglementations en matière d'emballage
Nos matériaux d'étiquetage destinés aux produits pharmaceutiques et de santé sont conçus pour répondre aux réglementations relatives aux emballages. Nos matériaux d'étiquetage sont conformes à toutes les exigences réglementaires et sécuritaires applicables en matière d'emballage et de contact alimentaire pour leur utilisation prévue ::
- Réglementations relatives aux emballages concernant les métaux lourds : directive européenne 94/62/CE et Toxics in Packaging Clearinghouse (TPCH), anciennement connue sous le nom de Coalition of North-eastern Governors (CONEG, États-Unis).
- La conformité des adhésifs aux réglementations européennes en matière de contact alimentaire ci-dessous a été évaluée :
- règlement-cadre (CE) n° 1935/2004 concernant les matériaux et objets destinés à entrer en contact avec des denrées alimentaires ; et
- règlement (CE) n° 2023/2006 relatif aux bonnes pratiques de fabrication des matériaux et objets destinés à entrer en contact avec des denrées alimentaires.
- Conformité à la réglementation de la FDA sur le contact alimentaire :
- FDA 21 CFR § 110 – Bonnes pratiques de fabrication (BPF)
Pourquoi utiliser des solutions d'étiquetage pharmaceutique dédiées ?
Les étiquettes transportent des informations médicales cruciales tout au long du cycle de vie du produit, tout en jouant également un rôle dans la protection et le scellage du contenu du produit. La durabilité, la qualité d'impression, les performances, la conformité aux réglementations et la sécurité des produits ne sont que quelques-unes des caractéristiques que l'étiquette doit posséder. En choisissant des solutions d'étiquetage pharmaceutique dédiées, les marques et les entreprises s'assurent que ces matériaux d'étiquetage sont :
- us et prétestés pour les applications pharmaceutiques ;
- développés en tenant compte de la sécurité des produits et de la conformité ;
- dotés d'une garantie de disponibilité et de stabilité à long terme, avec service de notification de gestion des changements et disponibilité assurée ;
- pris en charge par un service de documentation pour faciliter la validation réglementaire des produits par l'utilisateur final.